


 Sign Out
Sign Out

The most common serious adverse reactions were diarrhoea (2%) and hyperglycaemia (2%). Nine percent of patients permanently discontinued enfortumab vedotin for adverse reactions; the most common adverse reaction (≥2%) leading to dose discontinuation was peripheral sensory neuropathy (4%). Adverse reactions leading to dose interruption occurred in 44% of patients; the most common adverse reactions (≥2%) leading to dose interruption were peripheral sensory neuropathy (15%), fatigue (7%), rash maculo-papular (4%), aspartate aminotransferase increased (4%), alanine aminotransferase increased (4%), anaemia (3%), diarrhoea (3%) and hyperglycaemia (3%). Thirty percent of patients required a dose reduction due to an adverse reaction; the most common adverse reactions (≥2%) leading to a dose reduction were peripheral sensory neuropathy (10%), fatigue (5%), rash maculo-papular (4%) and decreased appetite (2%).
Tabulated summary of adverse reactions: The safety of enfortumab vedotin as monotherapy has been evaluated in 680 patients with locally advanced or metastatic urothelial cancer receiving 1.25 mg/kg on Days 1, 8 and 15 of a 28-day cycle in clinical studies (see Table 4). Patients were exposed to enfortumab vedotin for a median duration of 4.7 months (range: 0.3 to 34.8 months).
Adverse reactions observed during clinical studies are listed as follows by frequency category. Frequency categories are defined as follows: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. (See Table 4.)
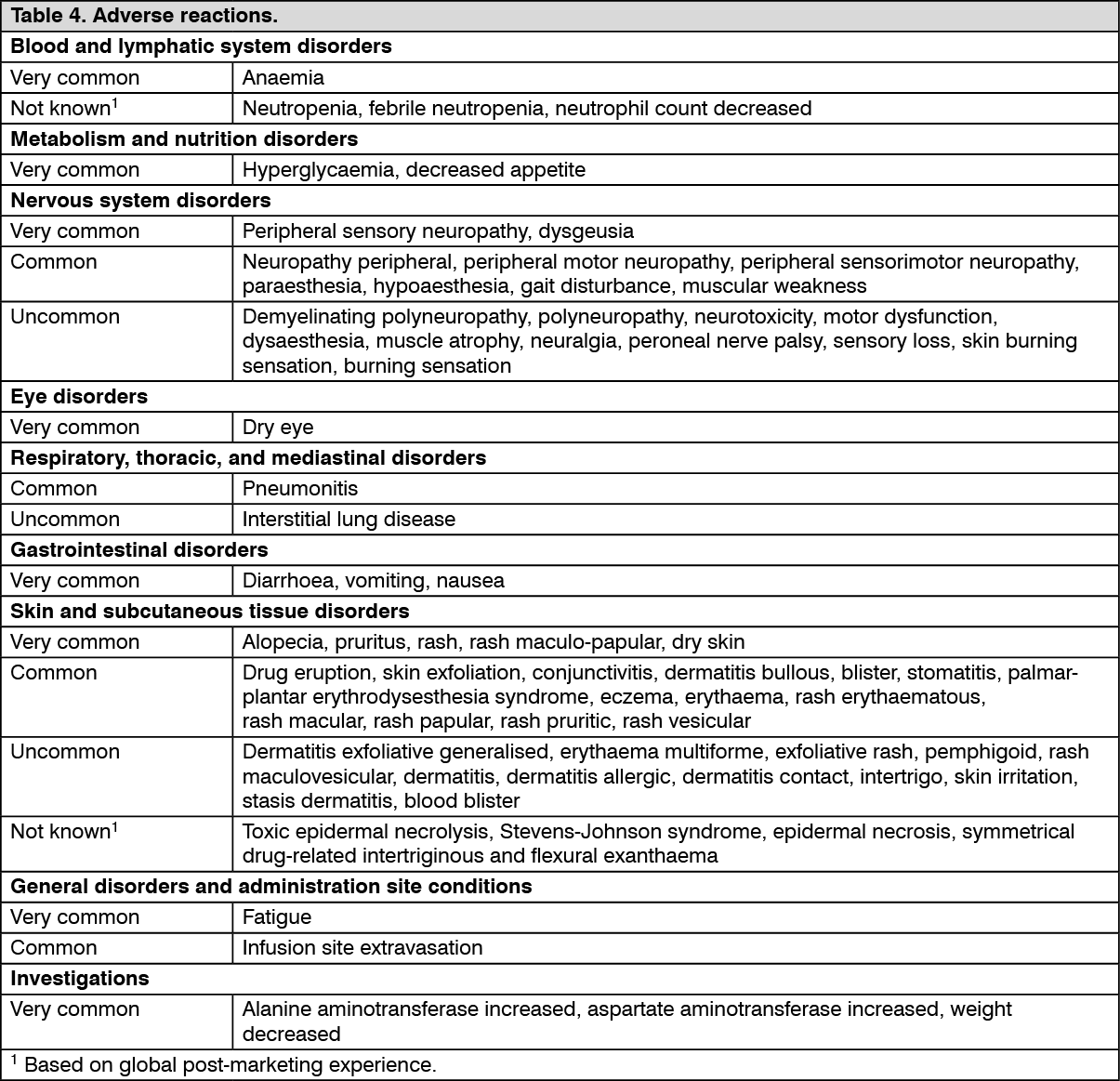 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Immunogenicity: A total of 590 patients were tested for immunogenicity to enfortumab vedotin 1.25 mg/kg; 15 patients were confirmed to be positive at baseline for anti-drug antibody (ADA), and in patients that were negative at baseline (N=575), a total of 16 (2.8%) were positive postbaseline (13 transiently and 3 persistently). Due to the limited number of patients with antibodies against PADCEV, no conclusions can be drawn concerning a potential effect of immunogenicity on efficacy, safety or pharmacokinetics.
Skin reactions: In clinical studies, skin reactions occurred in 55% (375) of the 680 patients treated with enfortumab vedotin 1.25 mg/kg. Severe (Grade 3 or 4) skin reactions occurred in 13% (85) of patients and a majority of these reactions included maculo-papular rash, rash erythematous, rash or drug eruption. The median time to onset of severe skin reactions was 0.62 months (range: 0.1 to 6.4 months). Serious skin reactions occurred in 3.8% (26) of patients.
In the EV-201 (N=214) clinical study, of the patients who experienced skin reactions, 75% had complete resolution and 14% had partial improvement (see Precautions).
Pneumonitis/ILD: In clinical studies, pneumonitis occurred in 15 (2.2%) and ILD occurred in 2 (0.3%) of the 680 patients treated with enfortumab vedotin 1.25 mg/kg. Less than 1% of patients experienced severe (Grade 3-4) pneumonitis or ILD. Pneumonitis or ILD led to discontinuation of enfortumab vedotin in 0.1% and 0.3% of patients, respectively. There were no deaths from ILD or pneumonitis. The median time to onset of any grade pneumonitis or ILD was 3.6 months (range: 0.8 to 6.0 months) and the median duration was 1.4 months (range: 0.2 to 27.5 months). Of the 17 patients who experienced pneumonitis or ILD, 6 (35.3%) had resolution of symptoms.
Hyperglycaemia: In clinical studies, hyperglycaemia (blood glucose >13.9 mmol/L) occurred in 14% (98) of the 680 patients treated with enfortumab vedotin 1.25 mg/kg. Serious events of hyperglycaemia occurred in 2.2% of patients, 7% of patients developed severe (Grade 3-4) hyperglycaemia and 0.3% of patients experienced fatal events, one event each of hyperglycaemia and diabetic ketoacidosis. The incidence of Grade 3-4 hyperglycaemia increased consistently in patients with higher body mass index and in patients with higher baseline haemoglobin A1C (HbA1c). The median time to onset of hyperglycemia was 0.6 months (range: 0.1 to 20.3).
In the EV-201 (N=214) clinical study, at the time of their last evaluation, 61% of patients had complete resolution, and 19% of patients had partial improvement (see Precautions).
Peripheral neuropathy: In clinical studies, peripheral neuropathy occurred in 52% (352) of the 680 patients treated with enfortumab vedotin 1.25 mg/kg. Four percent of patients experienced severe (Grade 3-4) peripheral neuropathy including sensory and motor events. The median time to onset of Grade ≥2 was 4.6 months (range: 0.1 to 15.8).
In the EV-201 (N=214) clinical study, at the time of their last evaluation, 19% of patients had complete resolution, and 39% of patients had partial improvement (see Precautions).
Ocular disorders: In clinical studies, 30% of patients experienced dry eye during treatment with enfortumab vedotin 1.25 mg/kg. Treatment was interrupted in 1.3% of patients and 0.1% of patients permanently discontinued treatment due to dry eye. Severe (Grade 3) dry eye only occurred in 3 patients (0.4%). The median time to onset of dry eye was 1.7 months (range: 0 to 19.1 months) (see Precautions).
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
View ADR Monitoring Form